마르판증후군 — 대동맥 기저부 확장·수정체 아탈구·긴팔다리·경막 팽윤 4가지 피브릴린-1 결함 유형
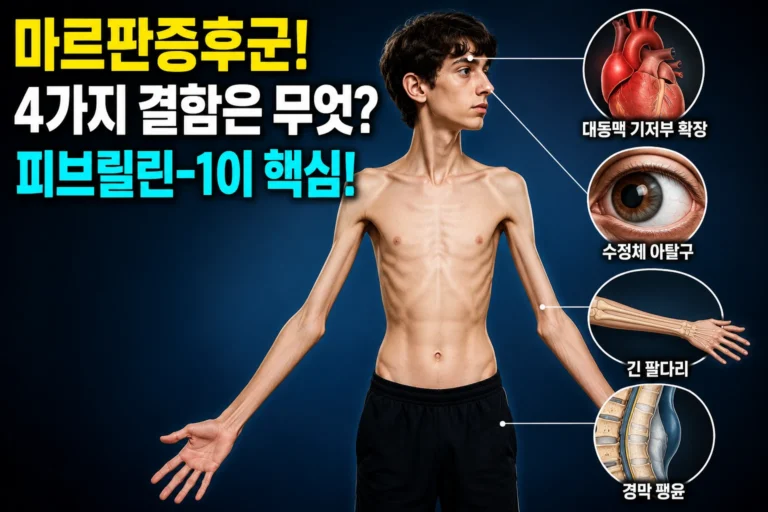
마르판증후군은 FBN1 유전자 이상으로 피브릴린-1 결합조직이 전반적으로 약해지는 선천성 유전 질환으로, 전 세계 유병률은 약 1/5,000이며 한국에서도 약 1만 명이 영향을 받는 것으로 추정됩니다.
대동맥 기저부 확장은 90% 이상에서 발생하고, 무치료 대동맥 박리는 시간당 1~2%의 사망률을 보이므로 직경 5cm 초과 시 예방적 수술을 고려해야 합니다.
키가 크고 팔다리가 길거나 수정체 아탈구·척추측만 등이 의심된다면 Ghent 기준에 따라 심초음파·안과·유전자 검사를 빠르게 받는 것이 치명적 합병증을 막는 핵심입니다.
FBN1 유전자 이상으로 피브릴린-1 결합조직이 약해지는 유전 질환. 유병률 1/5,000.
대동맥 박리. 무치료 시 시간당 1~2% 사망률. 직경 5cm 초과 시 수술 고려.
Ghent 기준으로 심초음파·세극등 검사·FBN1 유전자 검사를 조합해 판정.
격렬한 접촉 운동·중량 역기는 금지. 가벼운 유산소는 심장 전문의 상담 후 가능.
마르판증후군이란 — FBN1 유전자와 피브릴린-1 결합조직 결함
💡 한 줄 요약: 15번 염색체의 FBN1 유전자가 변이되면 피브릴린-1 단백질 구조가 무너지고, 전신의 결합조직이 동시에 약해지는 유전 질환입니다.
마르판증후군은 혈관·눈·뼈·폐·피부 등 결합조직이 있는 모든 부위에 영향을 미칩니다. FBN1 유전자가 정상적으로 만들어야 할 피브릴린-1 단백질이 결함을 가지면, 결합조직을 지지하는 미세섬유망이 취약해집니다. 그 결과 TGF-β 신호 경로가 과활성화되어 대동맥 벽, 눈의 섬모체소대, 뼈의 성장판, 척추 경막까지 폭넓게 손상됩니다.
15번 염색체에 위치. 2,700가지 이상의 변이가 보고됨. 25~30%는 새 돌연변이(de novo).
미세섬유 구조물이 정상 장력을 버티지 못함. 결합조직 전반의 탄성·강도 저하.
피브릴린-1이 TGF-β를 붙잡지 못해 신호가 폭주. 대동맥 중간층(tunica media) 분해 촉진.
유전 방식은 상염색체 우성입니다. 부모 중 한 명이 환자라면 자녀가 물려받을 확률이 50%에 달합니다. 나머지 25~30%는 부모에게 이상이 없어도 자연 발생한 새 돌연변이로 나타납니다. 전 세계 유병률은 약 1/5,000이며 성별·인종 구분 없이 발생합니다.
※ 참고 자료: Loeys BL et al., Journal of Medical Genetics, 2010 (Ghent nosology revised); OMIM FBN1
첫 번째 유형: 대동맥 기저부 확장 — 가장 위험한 심혈관 이상
💡 한 줄 요약: 마르판증후군 환자 90% 이상에서 대동맥 기저부가 확장되며, 직경 5cm를 넘으면 박리 예방을 위한 수술을 적극 검토해야 합니다.
대동맥 기저부는 심장 바로 위에서 혈압을 가장 직접적으로 받는 부위입니다. 피브릴린-1 결함으로 대동맥 중간층이 약해지면 혈압을 버티지 못하고 서서히 늘어납니다. 초기에는 거의 무증상이지만, 대동맥이 갑자기 찢어지는 박리가 발생하면 흉부와 등 사이에 찢어지는 듯한 극심한 통증이 나타납니다.
무치료 대동맥 박리는 발생 후 시간당 1~2%의 사망률이 보고됩니다. 갑작스러운 흉통·등 통증·혈압 불안정이 나타나면 즉시 응급실을 방문해야 합니다.
수술 결정 기준은 대동맥 직경 5.0cm입니다(ESC 가이드라인 2022). 가족 중 박리 병력이 있거나 확장 속도가 빠를 때는 4.5cm에서도 수술을 앞당기기도 합니다. 심초음파로 대동맥 기저부 직경을 매년 또는 6개월마다 추적 관찰하는 것이 핵심입니다. 대동맥 압력을 낮추기 위해 베타차단제나 ARB 계열 약물도 함께 사용합니다.
대동맥 질환에서 판막 문제도 함께 동반될 수 있으며, 대동맥 판막 협착 증상과 겹치는 경우도 있으므로 정확한 감별 검사가 필요합니다.

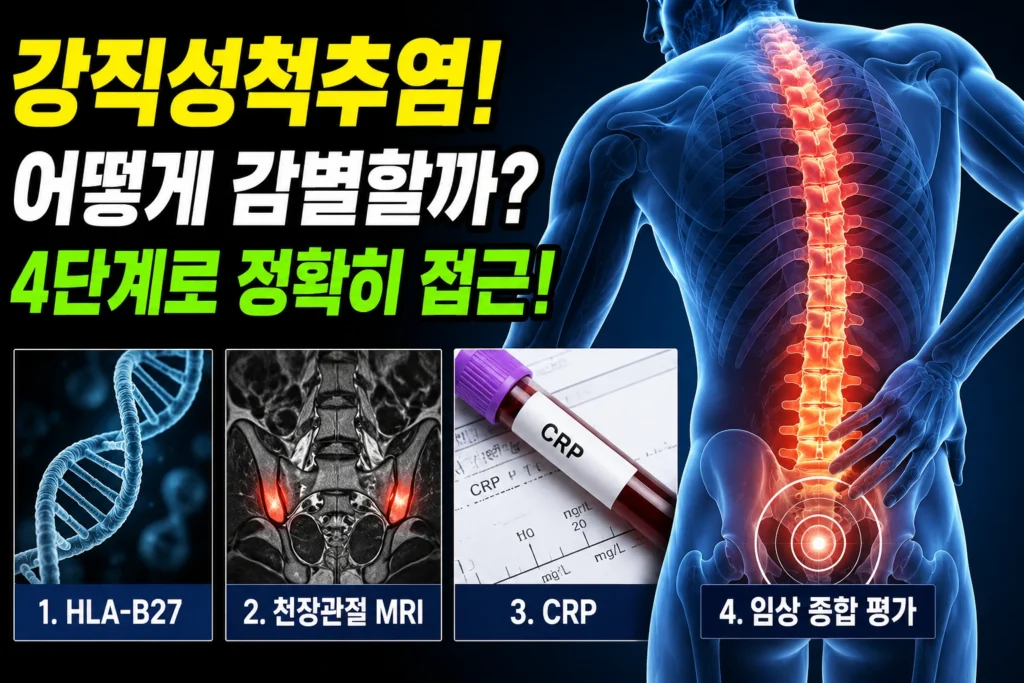
마르판증후군의 4가지 임상 유형 — 대동맥 기저부 확장, 수정체 아탈구, 긴팔다리·거미손가락, 경막 팽윤을 신체 부위별로 표시한 의학 인포그래픽
두 번째 유형: 수정체 아탈구 — 상방 탈구와 고도 근시
💡 한 줄 요약: 마르판증후군 환자의 60~70%에서 수정체를 고정하는 섬모체소대가 약해져 수정체가 위쪽으로 밀려나는 아탈구가 발생합니다.
눈 속 수정체는 섬모체소대라는 미세 섬유 다발로 매달려 있습니다. 피브릴린-1이 이 섬유의 주성분이기 때문에, 마르판증후군에서는 섬모체소대가 약해지고 수정체가 제자리를 벗어납니다. 탈구 방향은 대개 위쪽(상방)으로, 마르판과 달리 동형 시스틴뇨증에서는 아래쪽으로 탈구되는 점이 감별에 중요합니다.
3디옵터 이상의 근시가 흔하게 동반. 전신 점수 산정 시 1점으로 인정.
수정체 가장자리가 시축을 침범할 때 복시가 나타남. 빠른 안과 평가 필요.
Ghent 기준에서 수정체 아탈구 확인에 필수. 동공 산대 후 상방 탈구 여부 확인.
수정체 아탈구가 확인되고 FBN1 돌연변이(대동맥 관련 변이)가 함께 검출되면 그것만으로도 마르판증후군 확진이 됩니다. 수술 후에도 망막 박리 위험이 남아있으므로 정기적인 안과 추적이 필요합니다.
눈꺼풀이 처지거나 눈 주위 근육에 이상이 함께 생길 때는 안검하수 원인을 추가로 감별해 볼 필요가 있습니다.
※ 참고 자료: Ghent nosology, Journal of Medical Genetics, 2010
세 번째 유형: 긴팔다리·거미손가락·오목가슴 — 골격 이상 패턴
💡 한 줄 요약: 팔짱지수(팔 길이/신장) 1.05 초과, 엄지·손목 징후 양성, 오목가슴·척추측만이 전형적이며, 이를 수치화한 전신 점수 7점 이상이면 진단 기준에 부합합니다.
마르판증후군의 골격 이상은 뼈가 과도하게 길어지는 특성에서 시작됩니다. 피브릴린-1 결함이 성장판 주변 결합조직을 약화시켜 뼈가 정상보다 길고 가늘게 자랍니다. 키가 크고 손발이 긴 체형은 마르판증후군을 의심하는 첫 신호입니다.
팔 길이(손가락 끝 기준)를 신장으로 나눈 값. 1.05를 초과하면 비정상적으로 긴 팔.
주먹을 쥐었을 때 엄지손가락이 새끼손가락 쪽으로 완전히 튀어나옴. 전신 점수 1점.
한 손으로 반대쪽 손목을 감싸면 엄지와 새끼손가락이 겹침. 전신 점수 추가.
오목가슴(pectus excavatum) 2점, 새가슴(pectus carinatum) 1점. 폐 기능에도 영향.
20도 이상 척추측만 또는 척추전방전위 시 전신 점수 1점. 통증·신경 증상 유발.
pes planus(평발)와 동반된 발뒤꿈치 내반 변형. 전신 점수 2점으로 중요한 항목.
엄지 징후+손목 징후가 동시에 양성이면 전신 점수에서 3점을 얻어 진단에 유리합니다. 전신 점수가 7점 이상이고 대동맥 기저부 Z-score가 2 이상이면 Ghent 기준으로 마르판증후군에 해당합니다.
거미손가락(arachnodactyly)처럼 관절이 가늘고 길어 보이는 경우, 어린 나이에 관절 불편감이 동반된다면 소아 관절 이상 감별도 함께 검토해볼 필요가 있습니다.
※ 참고 자료: Ghent nosology revised, Loeys BL et al., 2010
네 번째 유형: 경막 팽윤 — 요통과 하지 방산통의 원인
💡 한 줄 요약: 마르판증후군 환자의 60~70%에서 척추 경막이 부풀어 주변 신경을 누르고, 반복되는 요통·하지 방산통·두통의 원인이 됩니다.
경막(dura mater)은 척수를 감싸는 강인한 막입니다. 결합조직 취약으로 경막이 약해지면, 뇌척수액 압력에 눌려 풍선처럼 팽창합니다. 이를 경막 팽윤(dural ectasia)이라고 하며 주로 요천추 부위에서 발생합니다. Ghent 전신 점수에서 2점을 차지할 만큼 진단 비중도 큽니다.
아침에 일어날 때 심해지거나 장시간 서 있으면 악화되는 특징이 있습니다. 원인 불명의 반복성 요통이 있다면 경막 팽윤을 감별해야 합니다.
CT나 일반 X선으로는 확인이 어렵고 MRI가 표준입니다. 경막 팽윤 자체보다 통증 관리가 목표이며, 심한 경우 수술적 강화술을 시행합니다.
경막 팽윤은 척추에 관한 불필요한 수술을 예방하는 데도 중요합니다. 마르판증후군 확진 전에 단순 추간판탈출증으로 오인되어 수술을 받는 경우가 있으므로, 젊은 나이의 반복 요통과 마르판 체형이 함께 있다면 전문가 감별이 필요합니다.
※ 참고 자료: Ghent nosology, Journal of Medical Genetics, 2010; ESC Guidelines 2022
마르판증후군 자가진단 체크리스트 7가지
💡 한 줄 요약: 아래 7가지 항목 중 3개 이상이면 전문의 상담을 권장하며, 5개 이상이면 Ghent 기준 정밀 검사가 필요합니다.
✅ 0~2개 해당
당장 마르판증후군을 강하게 의심할 근거는 낮습니다. 단, 키가 빠르게 자라는 청소년기에는 체형 변화를 주기적으로 관찰하고, 새로운 증상이 생기면 다시 체크해보세요.
⚠️ 3~4개 해당
여러 항목이 겹친다면 마르판증후군 관련 진료를 받아보는 것이 좋습니다. 심장내과 또는 유전질환 클리닉에서 심초음파와 안과 검사를 먼저 의뢰하세요.
🚨 5개 이상 해당
Ghent 기준 정밀 검사(심초음파·세극등·FBN1 유전자 검사)가 시급합니다. 대동맥 기저부 상태 확인이 최우선이며, 유전질환 전문 센터 방문을 권장합니다.
마르판증후군 임상 유형별 비교
💡 한 줄 요약: 대동맥 기저부 확장이 90% 이상으로 가장 흔하며 치명적이고, 수정체 아탈구·경막 팽윤은 각각 60~70%로 동반되며, 골격 이상은 거의 모든 환자에서 관찰됩니다.
| 유형 | 발생률 | 주요 증상 | 진단 검사 | 위험도 |
|---|---|---|---|---|
| 대동맥 기저부 확장 | 90% 이상 | 초기 무증상 → 흉통·등 통증·혈압 불안정 | 심초음파 (매년~6개월) | ⭐⭐⭐ 최고 |
| 수정체 아탈구 | 60~70% | 고도 근시, 복시, 시야 흐림 | 세극등(slit-lamp) 검사 | ⭐⭐ 중간 |
| 골격 이상 | 거의 전체 | 긴팔다리, 거미손가락, 오목가슴, 척추측만 | 전신 점수 산정·방사선 촬영 | ⭐ 낮음~중간 |
| 경막 팽윤 | 60~70% | 요통, 하지 방산통, 두통, 복부 불편감 | MRI 요천추 | ⭐ 낮음~중간 |
※ 참고 자료: Ghent nosology, Loeys BL et al., Journal of Medical Genetics, 2010; ESC Guidelines 2022
자주 묻는 질문 (FAQ)
Q
마르판증후군이란 정확히 어떤 병인가요?
▼
Q
마르판증후군은 반드시 유전되나요?
▼
Q
마르판증후군은 어떻게 진단하나요?
▼
Q
대동맥 박리는 어떤 증상으로 알 수 있나요?
▼
Q
마르판증후군 환자가 피해야 할 운동은 무엇인가요?
▼
Q
키가 크고 손발이 길면 마르판증후군인가요?
▼
Q
마르판증후군은 완치가 가능한가요?
▼
정리하며
마르판증후군은 FBN1 유전자 하나의 이상이 대동맥·눈·뼈·경막 전반에 걸쳐 파급되는 다계통 질환입니다. 대동맥 기저부 확장이 90% 이상에서 발생하며, 조기에 발견하지 못하면 대동맥 박리라는 치명적인 결과로 이어질 수 있습니다.
키가 크고 팔다리가 긴 체형에 엄지·손목 징후, 고도 근시, 반복 요통 중 두 가지 이상이 겹친다면 미루지 말고 심초음파와 안과 검사부터 받아보세요. 유전질환 전문 클리닉에서 Ghent 기준에 따른 정밀 평가를 통해 조기에 관리를 시작하는 것이 대동맥 박리를 예방하는 가장 확실한 방법입니다.
본 글은 건강 정보 제공을 목적으로 작성되었으며, 개인의 질환 치료나 의학적 결정에 적용하기 전에는 반드시 전문의와 상담하시기 바랍니다.